Zespół post-polio i stwardnienie boczne zanikowe – podobieństwa, różnice i dylematy diagnostyczne
prof. dr hab. n. med. Ewa Matyja; dr n. med. Milena Laure-Kamionowska
Zakład Neuropatologii, Instytut Medycyny Doświadczalnej i Klinicznej im. M. Mossakowskiego PAN, Warszawa
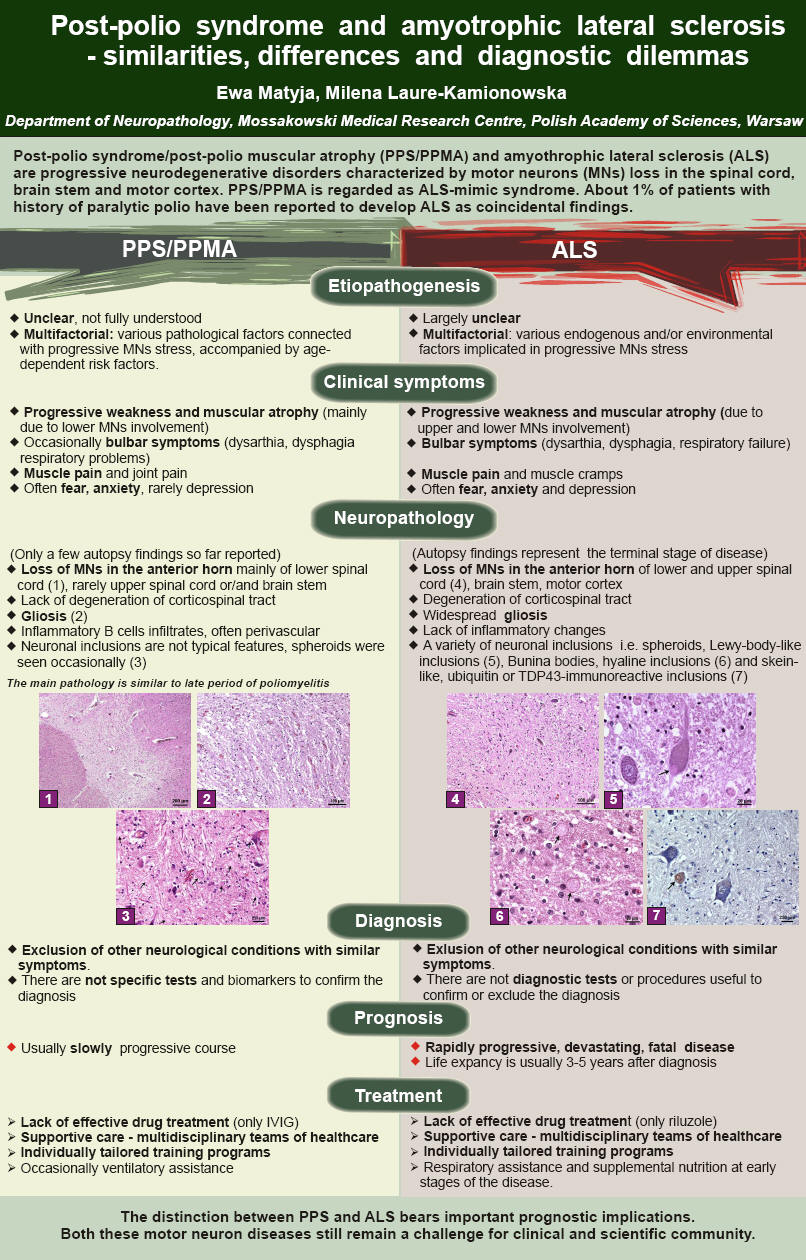
Zespół post-polio (PPS) i stwardnienie boczne zanikowe (SLA), stanowią schorzenia neurodegeneracyjne, które cechuje ubytek neuronów ruchowych (MNs) w rdzeniu kręgowym, pniu mózgu oraz korze ruchowej. Odróżnienie PPS i SLA niesie ważne implikacje terapeutyczne, ponieważ SLA stanowi dramatycznie rozwijającą się, śmiertelną chorobę z krótkim okresem przeżycia, podczas gdy PPS jest powoli postępującym zespołem chorobowym. Obie te choroby neuronu ruchowego (ang. motor neuron diseases – MNDs) pozostają wciąż wyzwaniem dla klinicystów i naukowców, i wymagają lepszego zrozumienia etiologii i skuteczności leczenia.
Objawy kliniczne, rozwijające się u pacjentów z zespołem post-polio z zanikiem mięśni (ang. post-polio muscular atrophy – PPMA), są podobne do początkowych objawów SLA i obejmują osłabienie i zanik mięśni, wskutek zajęcia dolnego i górnego neuronu ruchowego, czasem z towarzyszącymi problemami oddechowymi oraz zaburzeniami połykania. Zmiany neuropatologiczne w PPS i SLA są podobne i cechuje je ubytek MNs, występują jednak pewne różnice w obrazie histopatologicznym. Badania neuropatologiczne w PPS oparte są na nielicznych, dostępnych badaniach autopsyjnych. Wykazały one ubytek MNs w rogach przednich rdzenia kręgowego z towarzyszącą glejozą oraz naciekami zapalnymi z limfocytów B, bez zajęcia dróg korowo-rdzeniowych. W rodzinnej postaci SLA u ludzi obserwuje się obecność różnorodnych inkluzji neuronalnych, takich jak wtręty przypominające ciałka Lewy’ego, ciałka Buniny oraz wtręty hialinowe i ubikwityno-dodatnie, które nie są typowe dla PPS. Nacieki zapalne w przypadkach SLA obserwowane są sporadycznie.
Etiopatogeneza SLA i PPS nie jest do końca poznana i pod uwagę bierze się udział licznych czynników patologicznych, związanych z postępującym stresem MNs oraz towarzyszącymi czynnikami ryzyka, związanymi z wiekiem. Ponadto, u około 1% pacjentów z historią porażennego polio występuje koincydencja polio i SLA. Diagnoza histopatologiczna w takich przypadkach jest bardzo trudna, ponieważ zmiany typowe dla PPS i/lub SLA rozwijają się na podłożu zmian patologicznych w obrębie rdzenia kręgowego, pozostałych po pierwotnej infekcji poliomyelitis. Kryteria diagnostyczne PPS i SLA oparte są na wykluczeniu innych schorzeń o podobnej symptomatologii, ponieważ nie ma specyficznych testów i biomarkerów dla potwierdzenia diagnozy.
Podstawowe postępowanie terapeutyczne w przypadku PPS i SLA opiera się głównie na łagodzeniu objawów i wymaga indywidualnie zaplanowanego programu ćwiczeń oraz modyfikacji stylu życia. Jednak ze względu na różnice w naturalnej progresji obu tych chorób neuronu ruchowego, postępowanie w przypadku pacjentów z SLA musi być bardziej agresywne i obejmuje włączenie oddechu wspomaganego przy pomocy respiratora oraz uzupełniającego odżywiania we wczesnych stadiach choroby.
![]()
Słowa kluczowe: PPS SLA; diagnostyka różnicowa.